





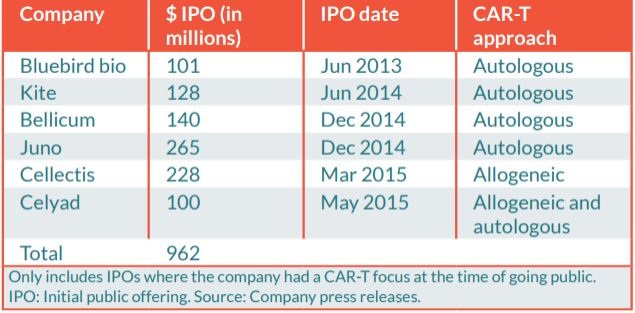
The biotech industry is diving into the field of highly individualized gene and cell therapy products. The first CAR-T therapy has just been approved in the US and other breakthrough technologies like mRNA and CRISPR have a foot in the door. But getting to the market is not straightforward, not only for developers but also for regulators, who should remain neutral toward the scientific and economic hype. Especially since for these therapies, side effects are often synonym of death. Still, the expectations on cell and gene therapies have never been this high: together, the IPOs of six CAR-T biotechs reached €800M in 2015.
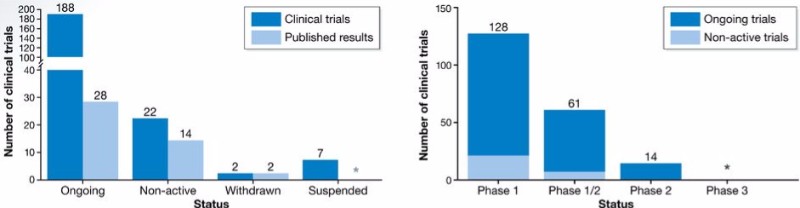
It is commonly accepted that pharmaceutical drugs take around 10 years to be developed. But CAR-T cells have taken about 30 years since the first development at the Weizmann Institute of Science in Israel. Today, dozens of CAR-T therapies are in clinical trials, but only two have been approved so far, both targeting the leukemia marker CD19. Why? One reason is that the regulatory pathways are challenging due to the unique nature and mechanism of action of CAR-T cells.
According to Kimberly Shultz from the Center for Biologics Evaluation and Research (CBER), manufacturing CAR-T cells is especially challenging because the product includes patient-specific cells that come in contact with many biological materials throughout the process. Therefore, having a proper regulatory strategy is essential to reach patients safely.
Sorting through European regulations
In Europe, CAR-T cells are defined as Advanced therapy medicinal products (ATMPs), and more precisely Gene Therapy Medicinal Products (GTMPs). It is mandatory to ensure this status before going further to avoid falling in between several regulatory pathways, such as that for Cell Therapy Medicinal Products.
The Committee for Advanced Therapy (CAT) reported so far more than 280 ATMP applications since 2009. A huge number compared to the few ATMPs products on the market but confirms that many products are on their way.
Quite the opposite for the certification procedure for SMEs, which has been adopted only 10 times in 8 years despite large promotion by the EMA. This procedure is one of the very few incentives dedicated to ATMPs, attesting quality of non-clinical data at early stages of development. It has been blamed for its complexity and disconnection between results and the rest of the regulatory pathway.
Although to my knowledge no specific certification procedures have been granted for CAR-T cell products so far, developers should reconsider the certification procedure for SMEs. For example, Celyad used this certification as a communication tool for C-Cure, its cardiovascular cell therapy, and could do the same with its CAR-T pipeline. I am sure that investors would be receptive to this EMA certification even if they have limited knowledge of the procedure.
Another relevant pathway for CAR-T cells just celebrated its 1 year anniversary with more success. The PRIME pathway was launched by the EMA to support the development of medicines that target an unmet medical need, which is the case of many CAR-T cells. So far, it has received more than 100 applications, a third of which were for ATMPs. Four CAR-T cell products from three pharmaceutical companies (KTE‐C19 from Kite Pharma; CTL019 from Novartis; JCAR015 and JCAR017 from Juno Therapeutics) have already been granted access to PRIME.

To help navigate all the options, it is possible to get advice from the CAT through its scientific advice procedure for ATMPs, which is a formal Q&A. However, this needs to be well prepared. Getting the right answer for an open question is a tough exercise.
Otherwise, it is very difficult to get informal feedback from regulatory agencies. Most of the time they are “shy” to answer by email, which could be used against them later in the regulatory process. And the angencies themselves often have weaknesses. When you take a look at the CVs of members of the CAT you realize quickly that most of them have a lot of experience with cell and gene technologies but limited experience within the industry.
To bridge the skill gap they often count on specialists for punctual assessments or before drafting new guidelines. For example, the EMA broadcasted a CAR-T workshop in 2016, where experts from academia, industry and regulatory authorities gathered to discuss how to facilitate the development of cancer treatments based on CAR-T cells.
Europe is currently working on creating common guidelines on good manufacturing practices (GMP) specific for ATMPs. New guidelines regarding clinical trial requirements for ATMPs will also be published by the end of 2018 to reduce current discrepancies across the EU. This will especially help trials run in multiple EU countries, which is currently a big challenge for CAR-T cell therapies, and especially in France, which developers consider slow in processing trials requests.
By 2020, a few other new ATMP guidelines will be published. However, guidelines covering specifically the field of CAR-T cells have not yet been planned in Europe, indicating how difficult it is to keep track of these rapidly evolving technologies.
Going international
Being limited to European approval would be a major mistake. In fact, less than 10% of the ongoing CAR-T clinical trials are being run here. All over the world, authorities are reviewing their guidelines, and China in particular has reached the largest number of CAR-T cell trials despite it started developing the technology later than North America.
The US FDA has made substantial progress in the past decade to address the rapidly evolving technologies needed for CAR-T manufacturing. Only in 2017, it generated 5 new guidance documents on cell and gene therapies. And Novartis’ CAR-T therapy Kymriah was approved through the FDA’s fast track designation.
The FDA is currently working on more specific guidance for technologies such as stem cell therapies, though we don’t have any timeframe for their release yet. But even more interestingly, it wants to create two unique databases that will proactively gather safety and manufacturing data for CAR-T cells targeting CD19.
One can hope that these new guidelines will give more practical input than current ones, which constantly repeat common knowledge regarding case-by-case approaches, iterative discussions with authorities and the mandatory exchange of information between scientists, physicians, developers, and regulators. At the end, it doesn’t help with major technical issues. Biotechs end up without an established pipeline for clinical translation like those used to approve small molecules or antibodies, so they have to rely on academic knowledge instead. As a result, so far only 20% of CAR-T cell trials are sponsored by the pharmaceutical industry.

This makes it very difficult to bridge the gap between early- and late-stage data. Novartis’ Kymriah, which has been mainly developed by academics, had to be built on years of experience to insure reproducibility, flexibility and a validated manufacturing process. Let’s hope that the fate of Kymriah won’t be the same as Dendreon’s Provenge — one of the very first ATMPs to be authorised, but withdrawn in 2015 — which was produced in the same facilities as Kymriah before Novartis bought them from Dendreon.
The MD Anderson Cancer Center recently published a paper titled Guidelines for handling CAR T cell side effects that gives an overview of how to address toxicity. Thanks to these initiatives, publications and trials, a gold standard for validation that could be reasonably assimilated into guidelines is finally emerging.
Challenges ahead
There are still many challenges that need to be properly addressed when developing CAR-T cells. These include viral vector testing, establishing GMP-compliant manufacturing, and demonstrating clinical equivalence between batched despite the inherent variability of the product. The EU CARAT consortium is currently working on identifying essential obstacles for the clinical development of CAR-T cells with the aim of publishing suggestions on how to translate research into clinical use.
Recently, Europe and the US have agreed to recognize each other’s inspections of manufacturing sites. But there are still large discrepancies between requirements among regions. For example, donor screening and testing, traceability and labeling, patient confidentiality, and best practices for apheresis can vary widely. Just the definition of starting material and raw material changes everything, since there are huge differences in quality control regarding the status of the material. This becomes particularly challenging when the starting material and final product are shipped across international borders.
Regulations on gene and cell therapy products have evolved over time and have relatively improved. But regulators all over the world must remain proactive and consider all possible limitations and challenges of CAR-T development in order to create proportional regulatory requirements for the benefit of patients. Either too permissive or too constrained frames could be disastrous.
CAR-T cell therapy could bring significant benefits to patients, and it is clear that regulators are keen to make these game-changing therapies available. This provides a great opportunity to create a common ground between authorities and improve borderless cooperation. Until we achieve harmonization, we’ll have to keep dealing with uncertainty and subjective regulatory decisions.
Images via aurielaki /Shutterstock; MJ Smith et al. (2016) Cell & Gene Therapy Insights; J Hartmann et al. (2017) EMBO Molecular Medicine, e201607485; EMA; BL Levine et al. (2017) Mol Ther Methods Clin Dev. 4: 92–101