





Current treatments for cystic fibrosis are not suitable for all patients and have a limited effect on this life-threatening disease. But new advances in the field promise to overcome these hurdles.
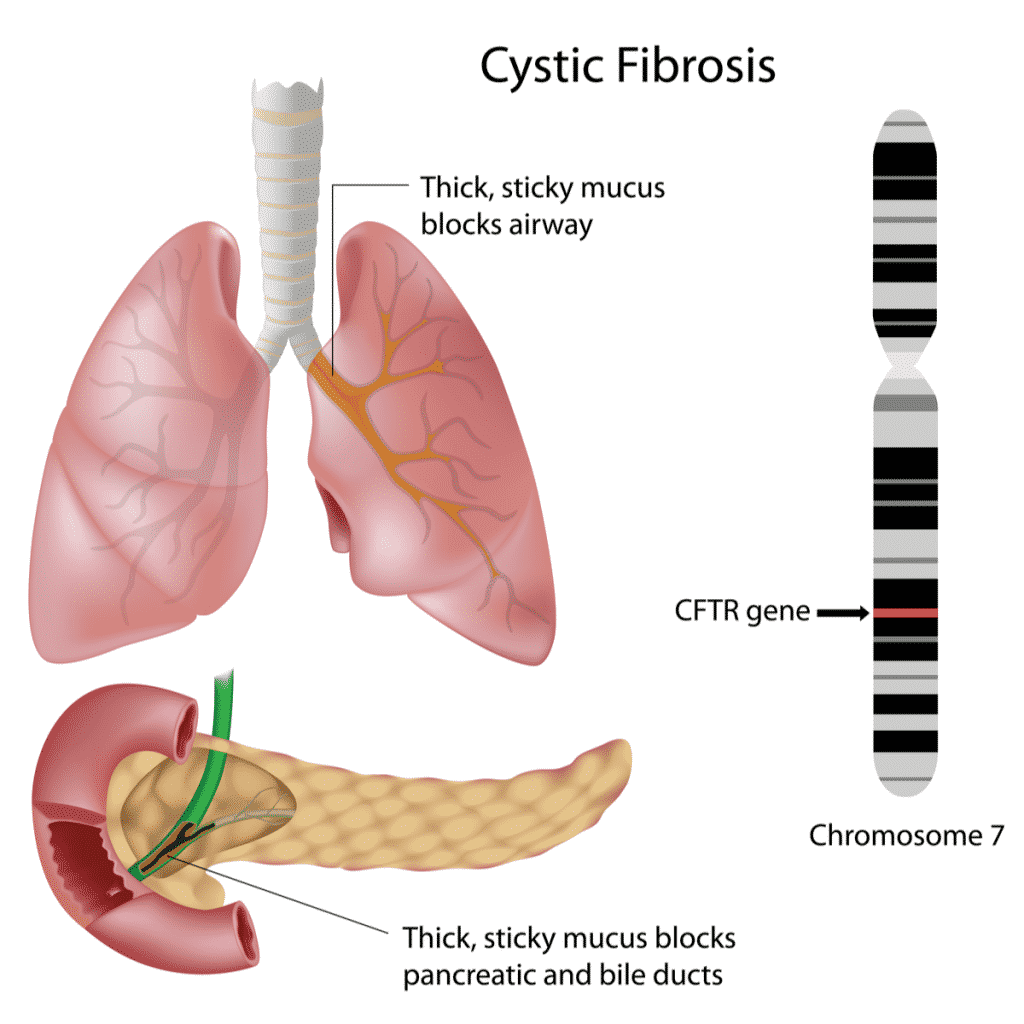
The cause of cystic fibrosis is very straightforward. Its treatment, however, is not. People diagnosed with cystic fibrosis have a mutation in a gene called CFTR. This gene encodes a protein that is responsible for transporting chloride to the surface of cells. Without chloride to attract water, the mucus that surrounds the cells in many organs becomes thick and sticky.
The main organ affected is the lungs, where the mucus clogs airways and makes them prone to infection and inflammation. The function of the pancreas, liver, and bowel is also affected — a single mutation ends up damaging the whole body. Currently available treatments are still not able to fully address the complexity of the disease.
“In the last 10 years there has been highly significant progress in the development of small molecules for treating cystic fibrosis,” said Deborah Gill, Professor at the University of Oxford. “The problem is that the most success has been observed in cystic fibrosis patients with only specific classes of mutation, and since there are nearly 2,000 different mutations in six different classes, many patients still have no treatment.”
“The average life expectancy of a cystic fibrosis patient, although improving, is approximately 40 years,” said John Ford, CEO of Enterprise Therapeutics, a UK biotech company developing drugs for respiratory diseases.
“New treatments are urgently needed for cystic fibrosis patients who do not benefit from CFTR repair drugs. In addition, patients on CFTR drugs are still experiencing long-term lung function decline – this needs to be addressed via alternative drug mechanisms.”
Gene therapy: going after the main culprit
The gene that causes cystic fibrosis was discovered in 1989, revealing for the first time that the disease is caused by mutations in a single gene.
“When the gene was discovered everyone thought that gene therapy for cystic fibrosis should be straightforward, but even though the discovery was 30 years ago, there is still no gene therapy available to cystic fibrosis patients,” said Gill.
“Not long after the discovery of the cystic fibrosis gene there were many research groups and biotech companies interested in developing a gene therapy. After many clinical trials showed that none were successful in cystic fibrosis patients, many groups lost interest.”
Gill, however, has not given up on this goal. She is part of a British consortium where the Universities of Oxford, Edinburgh, and University College London work together in the development of a gene therapy for the condition.
“If an effective gene therapy could be developed and given to children and young adults with cystic fibrosis, we would expect that they would be protected from developing lung disease, which is usually the fatal consequence of cystic fibrosis,” Gill explained.

The consortium has run several phase I/IIa proof-of-concept clinical trials, with the first one revealing in 2015 that the lung function of those patients receiving their gene therapy did not decline as much as it did for patients receiving a placebo. It was the first time a gene therapy showed clinical benefit in people with cystic fibrosis.
However, the improvement was moderate, so the researchers decided to modify the gene therapy to increase its efficacy. While the original version made use of tiny fat droplets to carry the DNA, the new one makes use of a hybrid virus.
The researchers have modified a monkey lentivirus — a kind of virus that can insert genetic material in the host cell — to carry the external ‘targeting’ proteins of a virus that naturally infects the lung.
The new therapy has been tested in a preclinical setting, where it has proved to be more effective than the previous version. In 2018, the consortium got funding from Boehringer Ingelheim, which has now exercised its license option to develop and commercialize the gene therapy.
“The next step is to test it in the lungs of cystic fibrosis patients,” said Gill.
The major challenge that led most gene therapies for cystic fibrosis to fail in the past is delivering the therapy to the lungs. To address this the consortium is now focusing on manufacturing the virus in large enough amounts to start clinical trials, a task taken on by the biotech company Oxford Biomedica.
“The lung is a large organ,” said Kyriacos Mitrophanous, CSO of Oxford Biomedica. “Normally with gene therapy, you are delivering small amounts to the brain or the eye, but for the lung, you will need a considerable amount of viral vectors, especially if you’re going to nebulize it because maybe half of the vector will be lost just from the nebulization and, obviously, breathed out.”
If the therapy proves able to modify enough lung cells, the partners expect the therapy would only need to be given every three to five years, or even just once. It would also remove the need for any other medications for lung function.
“Unlike the biotech approach where you do a mutation-specific therapy, this therapy would work in all cystic fibrosis patients,” explained Mitrophanous. “We’re putting back the correct version of the gene irrespective of whatever mutation the patient has.”
While few research groups are working on gene therapy for cystic fibrosis, a new generation of genetic treatments might soon emerge thanks to CRISPR-Cas9 gene editing. With this technology, it would be possible to revert mutations directly in the CFTR gene, although this kind of treatment would need to be tailored to each specific mutation causing cystic fibrosis.
For example, in early 2020, researchers of the Hubrecht Institute in the Netherlands used CRISPR base editing to repair CFTR mutations in vitro in the cells of cystic fibrosis patients, while keeping the rest of their genetic code intact.
The Swiss gene-editing company CRISPR Therapeutics and US firm Vertex Pharmaceuticals established a partnership back in 2015 to target cystic fibrosis using CRISPR-Cas9. However, this gene-editing technology has yet to be proven in humans, and research to apply it to cystic fibrosis is still at a very early stage.
Next-generation drugs: improving efficacy
As gene therapy has historically struggled to bring benefits to people living with cystic fibrosis, many other companies have turned to developing next-generation drugs for cystic fibrosis that are not restricted to specific mutations.
Among them is UK biotech Verona Pharma, a clinical-stage company developing a first-in-class drug that, according to former CEO Jan-Anders Karlsson, “acts as both a bronchodilator and an anti-inflammatory agent in a single molecule.”
The drug, called ensifentrine, blocks the activity of two enzymes involved in inflammation and is aimed at all cystic fibrosis patients independent from the mutation they carry. In addition, it has been shown to activate rare mutations of the CFTR protein, reducing the amount and thickness of the mucus.
“The anti-inflammatory effect may be particularly helpful to cystic fibrosis patients as its activity is independent of any particular gene mutation and there is no effective anti-inflammatory agent specifically approved for use in cystic fibrosis to date,” Karlsson said.
The company has already tested the effects of the drug in a phase II trial. The results showed that the patients’ lung function improved after a single dose of nebulized ensifentrine.
While the treatment would not be a cure, it has the potential to treat the symptoms and improve the quality of life for people with cystic fibrosis. The treatments patients currently receive aim to repair the defects caused by specific mutations in CFTR proteins, but their effect can be limited and lung inflammation and infection are not uncommon.
“CFTR repair does not treat the over 10% of cystic fibrosis patients with nonsense mutations and does not cure cystic fibrosis. In addition, many patients eligible for CFTR repair therapy do not benefit from these therapies,” said Ford.
His company, Enterprise Therapeutics, is developing drugs targeting the excess mucus that causes the airways to become blocked.
“Reducing mucus congestion will reduce the frequency of lung infections and improve patient quality of life,” explained Ford.
In particular, the company targets ion channel proteins that control fluid secretion and mucus clearance from the airways. In June 2021, it dosed the first subjects in a phase I trial for its lead cystic fibrosis therapy candidate.
Looking ahead: dealing with a complex disease
It is important to note that while many new approaches in development for cystic fibrosis bring hope to improve the lives of people with this condition, most of these efforts are limited to the effects that the disease has on the lungs.
While lung complications are the most severe symptom of cystic fibrosis, medicine is starting to look more closely at the effects cystic fibrosis has on other organs, such as the bowels, pancreas, and liver.
At the University Medical Center (UMC) Utrecht, in the Netherlands, researchers are working on the development of miniature organoids that replicate the structure of different tissues using cells from patients with cystic fibrosis.
This technology could yield results that are much more accurate at predicting how the human body will behave under different conditions and how it will react to experimental drugs. It could also help patients with rare forms of cystic fibrosis be prescribed a personalized treatment.
In 2019, the group published a study where 24 cystic fibrosis patients were prescribed personalized treatments after testing different drugs on intestinal organoids made from their cells.
“For the first time, this study demonstrated just how reliable such predictions can be for individual patients,” stated Jeffrey Beekman, who led the study. The UMC is currently working on a similar study, which is investigating 500 patients with rare forms of cystic fibrosis.
There is still a long way to go before this kind of practice becomes part of the standard care for patients with cystic fibrosis. But as scientists learn more about the disease’s multiple levels of complexity, the future is looking brighter for patients with the condition.
Much as the treatment of cystic fibrosis has considerably improved in the last decade, experts believe that the treatments currently being developed have the potential to revolutionize the lives of people with this debilitating disease still further in years to come.